Article Title: MhGeneS: An analytical pipeline to allow for robust microhaplotype genotyping
Authors: Julia C. Geue, Peng Liu, Sonesinh Keobouasone, Paul Wilson & Micheline Manseau
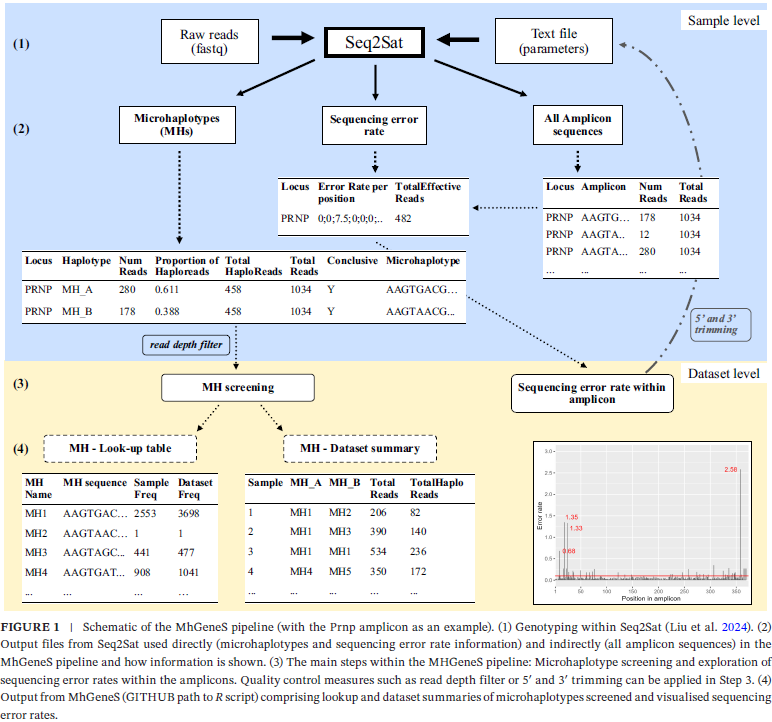
Abstract: Microhaplotypes are small linked genomic regions comprising two or more single-nucleotide polymorphisms (SNPs) that are being applied in forensics and are emerging in wildlife monitoring studies and genomic epidemiology. Typically, targeted in non-coding regions, microhaplotypes in exonic regions can be designed with larger amplicons to capture functional non-synonymous sites and minimise insertion/deletion (indel) polymorphisms. Quality control is an important first step for high-confidence genotyping to counteract such false-positive variants. As genetic markers with higher polymorphism compared to biallelic SNPs, it is critical to ensure sequencing errors across the microhaplotype amplicon are filtered out to avoid introducing false-haplotypes. We developed the MhGeneS pipeline which works in tandem with Seq2Sat to help validate microhaplotype genotyping of the coding region of genes, with broader applicability to any microhaplotype profiling. We genotyped microhaplotype regions of the Zfx (≅ 160 bp) and Zfy (≅ 140 bp) genes, as well as an exon of the prion protein (Prnp) gene (≅ 370 bp) in caribou (Rangifer tarandus) using paired-end Illumina technology. As important quality metrics affecting microhaplotype calling, we identified the sequencing error rate profile related to the overlap or non-overlap of paired-end reads as well as the read depth as significant. In the case of Prnp, we achieved confident microhaplotype calling through MhGeneS by removing small sections of the 5′ and 3′ amplicons and using a minimum read depth of 20. Read depth and sequence trimming may be locus-specific, and validation of these parameters is recommended before the high-throughput
profiling of samples.
Click here to access the full publication.



